Error Analysis#
Assessing statistical convergence and uncertainty in time-dependent simulations is essential for reliable conclusions. CRISP provides robust tools for error analysis that go beyond simple block averaging to offer statistically principled approaches.
Overview#
Block averaging divides trajectories into fixed-size blocks but is sensitive to block size choice. Autocorrelation function (ACF) analysis offers a more principled approach to quantify statistical inefficiency and evaluate convergence.
CRISP’s implementation removes the need to manually determine block size and enables straightforward error estimation through autocorrelation analysis.
Basic Error Analysis#
from CRISP.simulation_utility.error_analysis import error_analysis
import numpy as np
# Load MD trajectory energy data
data = np.load("energy.npy")
# Perform comprehensive error analysis
res = error_analysis(data)
# Display results
print(res["acf_results"])
print(res["block_results"])
Case Study: Zeolite MFI Chemical Shift Analysis#
This example demonstrates error analysis of 27Al chemical shifts in zeolite MFI from 1 ns molecular dynamics trajectories based on the work of Willimetz et al. [1].
MD Energy Convergence Analysis#
We examine statistical convergence by analyzing total energy standard error over different trajectory lengths using both ACF and block averaging:
from CRISP.simulation_utility.error_analysis import error_analysis
import numpy as np
# Load MD trajectory energy data
energy_data = np.load("energy.npy")
# Analyze different trajectory lengths
trajectory_lengths = [1000, 10000, 100000, 1000000] # in fs (1ps, 10ps, 100ps, 1ns)
for length in trajectory_lengths:
subset_data = energy_data[:length]
res = error_analysis(subset_data)
acf_error = res["acf_results"]["acf_err"]
block_error = res["block_results"]["block_err"]
print(f"Time: {length/1000:.0f} ps | ACF Error: {acf_error:.1f} kJ/mol | Block Error: {block_error:.1f} kJ/mol")
Results:
Time: 1 ps | ACF Error: 64.3 kJ/mol | Block Error: 8.7 kJ/mol
Time: 10 ps | ACF Error: 31.7 kJ/mol | Block Error: 6.9 kJ/mol
Time: 100 ps | ACF Error: 4.6 kJ/mol | Block Error: 3.4 kJ/mol
Time: 1000 ps | ACF Error: 0.8 kJ/mol | Block Error: 1.2 kJ/mol
Standard error decreases with increasing simulation length. At 1 ns, the SEM falls below 1 kJ/mol, indicating good sampling. ACF estimates are consistently more sensitive than block averaging, especially at shorter timescales, because ACF explicitly accounts for temporal correlations.
Chemical Shift Error Analysis#
# Predicted chemical shift for each frame
data = np.loadtxt("Al_nmr_results.txt", usecols=3, skiprows=1)
res = error_analysis(data)
print(res["acf_results"])
print(res["block_results"])
The autocorrelation time for chemical shift is approximately 50 fs, indicating rapid equilibration. Error is less than 0.05 ppm for both methods, suggesting 1 ns simulation is sufficient for reliable 27Al chemical shift prediction.
Visualization:
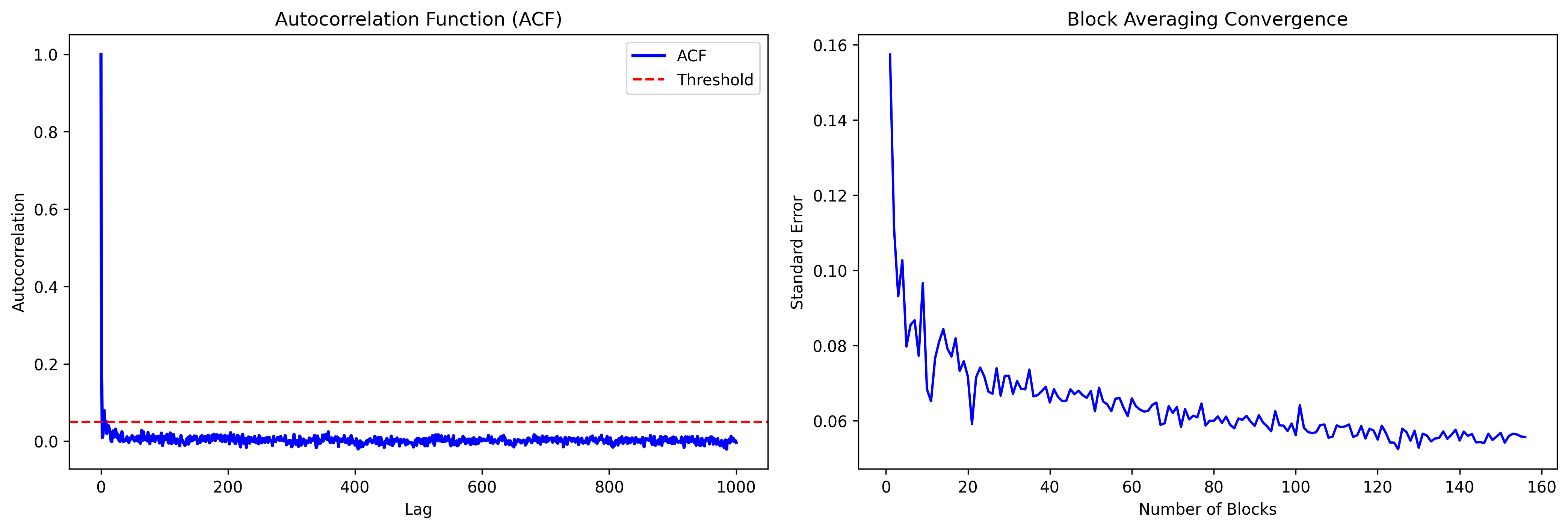
The plots show autocorrelation function decay and error convergence with simulation time.